Summary
Using fastqc, HISAT2, featureCounts, samtools, and Trimmomatic, an alignment pipeline was made for RNAseq data to the Chinese hamster ovary (CHO) genome. The annotation files were taken from NCBI. The workflow until the featureCounts step show’s an older annotation file. Note that the workflow is identical for the updated annotation file, but the updated annotation file provides higher alignments for the final output counts matrix.
Background
Raw files for the Transcriptomic data was taken from NCBI’s GEO datasets. The exact steps for aligning the raw RNAseq output can be seen below
- Download an SRA database file (or receive from Illumina sequencer device)
- Use the SRA toolkit to convert the SRA database file to a fastqc file
- Trim the fastqc file and run a quality check using Trimmomatic
- Use samtools to convert the fastqc file to bam file
- Sort the bam file using samtools
- Download the CHO genome sequences and annotation files
- Create genome indexes using HISAT2
- Get the counts matrix using featureCounts, the genome annotation, and the bam file.
- Parse out the counts from the TSV file output
The following is a workflow of building the genome indexes up until the final tab-separated-values (TSV )output. The SRA toolkit steps are not included In the following for two reasons: they were converted before making this portfolio, and Illumina devices often output the FASTQ file from sequencing runs.
Building Genome Indexes from Cricetulus Griseus genome
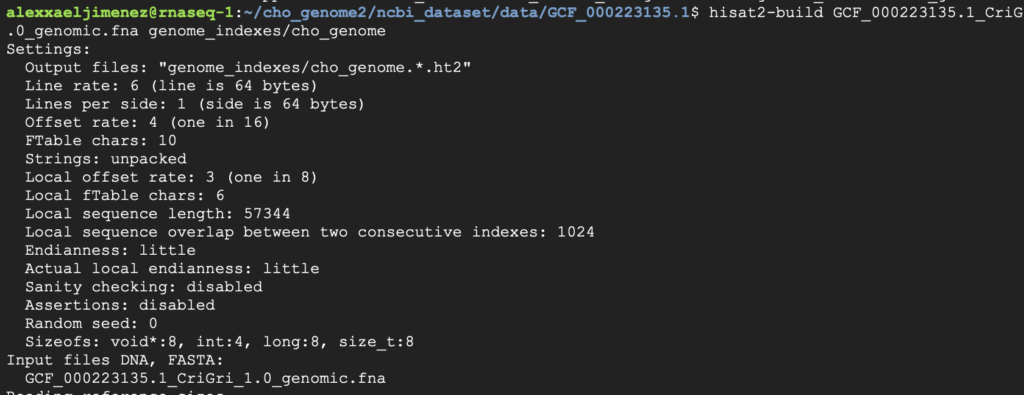
The final output after this command has run is 8 genome indexes. These genome indexes are crucial for HISAT2 to align the FASTQ files.

HiSAT2 for Aligning FASTQ Files to Cricetulus Griseus Genome
So it’s crucial to understand the protocol that our reads were generated under. Since this data was not generated by me, I’ll refer to the official sequence read archive for my data.

Given the data is paired and cDNA, the following command is appropriate for aligning the reads. Note that if you do run a command that doesn’t take into account the paired nature of this data, you’ll get an error or close to 0% alignment.

Here the output file was designated as a SAM file. Given the large size of these files, a BAM file, the compressed equivalent of a SAM, is often a better choice, especially in a cloud computing environment to minimize storage costs. Storage costs can be significant for a company constantly running its sequencer. Here is a summary report of the final alignment showing a successful alignment.

Sorting the SAM/BAM files
Sorting the SAM/BAM file is mainly for efficiency and speed for any other downstream tasks that might happen: featureCounts, and variant calling.

FeatureCounts For Creating Counts Matrix
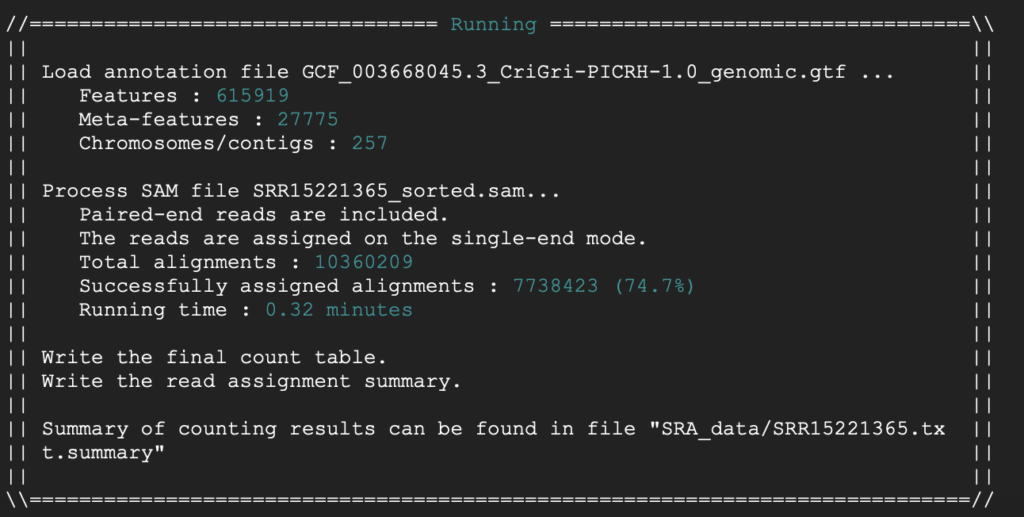
The following is a summary using the following genome assembly submission from NCBI.
High Level Programming Language (Python) for Further Analysis
Once the final counts matrix has been created, a higher-level programming language such as R or Python can be used for further analysis. R is a popular programming language in the biologist realm, but I prefer Python due to its broad package ecosystem in any domain. Python lags behind R slightly in the biotechnology world, but not by much due to its vast community. Some popular packages for analyzing RNAseq data include SCANpy, GSEApy, and other typical data science packages such as scikit-learn.
Future Improvements
If the protocol for read generation is not changing i.e. this is an in-house workflow for a pharmaceutical company, then the workflow can be automated using bash scripting and a higher-level programming language like Python. If Python is the language of choice, one can leverage the subprocess module to communicate directly with the bash scripts. Using AWS, this workflow could be further automated with the following services so that any data generated from a sequencer could be processed seamlessly.
- AWS S3 Storage Bucket for Raw Data and Event Notifications for Data Updates
- AWS ECS to Create a Docker App to Execute Bash Script with Python Front End for AWS RDS connection.
- AWS Relational Database Service (RDS) for final storage of output data.