Source Code: GitHub
Motivation Behind the Project
The inspiration for this project is from the following paper: Junction Tree Variational Autoencoder for Molecular Graph Generation by Jin et al. I wanted to recreate the work done by Jin et al to explore junction trees, chem-informatics, and test my coding abilities. The outcome of the decomposition is not identical to Jin et al’s representation, but that’s because I wanted to do this from scratch and likely some design decisions differed. This package could be used in the future as a feature-rich input for machine learning. I see some major advantages to this type of decomposition for Transformers.
Math Decomposition
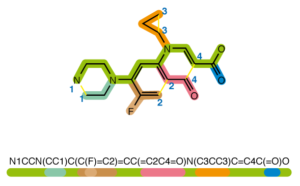
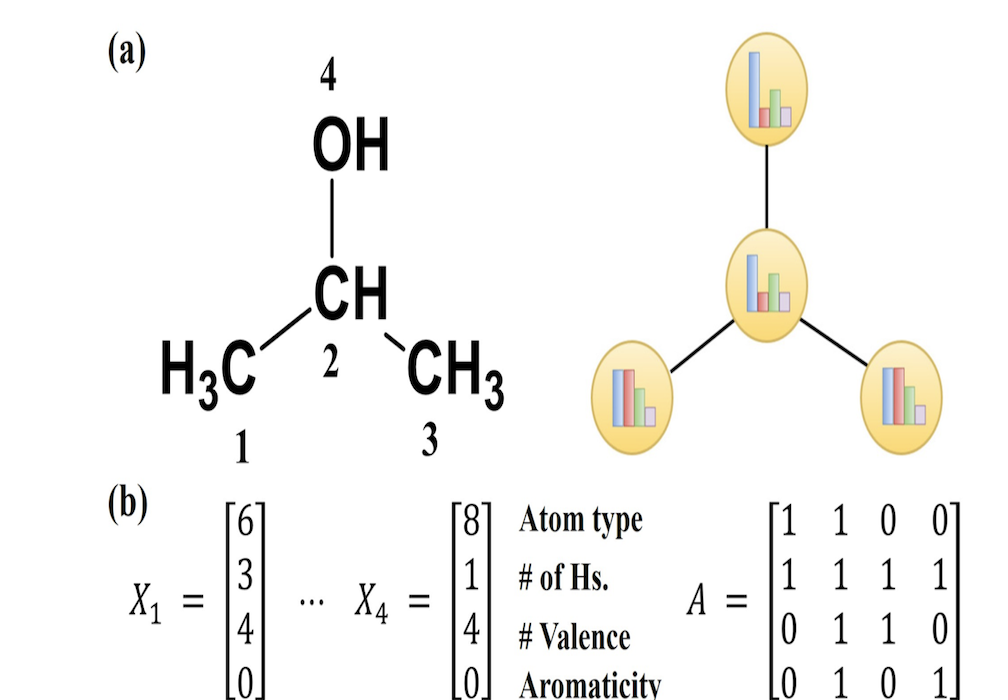
This project is a Python package that takes chemical SMILES and encodes them into a graph structure using a variety of chemical properties and preprocessing techniques below is an example of what a SMILE projection of a molecule might look like. Background on SMILES can be found here.
The target format of our data looks much different than our SMILES representation. Before we show what this format will look like, here is a list of properties used to decompose a molecule into its unique aspects.
Chemical Properties
- Aromaticity/Rings
- Bond Type (Single, Double, Triple)
- Chirality
- Tertiary/Quaternary Atoms
- Bond Isomerism (Cis/Trans/None)
- Valence
The molecule was than composed into a junction tree using the methodology used in Jin et al. The following is an image of the algorithm written by Jin et al.

A junction tree of the molecule pairs relevant groups of a molecule together. In this case, we define V2 as the ring structures of a molecule, which are important since they can be rigid and provide lots of information about their steric properties. V1 is defined as the bonds that do not belong to any ring. V0 is the intersection of three or more clusters, which in the case of a carbon atom, represents tertiary and quaternary carbons. This is important, because chirality, functional groups, and resonance structures can be around these points. These are then composed into a junction tree. An example of a junction tree can be seen below, where image (a) could represent the original connectivity of our molecule and image (b) represents the maximum spanning tree of our molecule. More information on maximum-spanning trees can be found here.
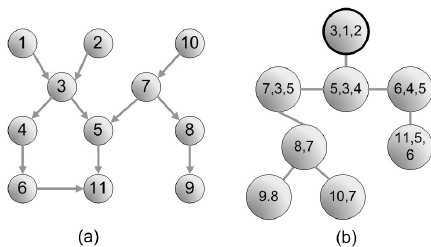
Applying this methodology and doing some additional preprocessing eventually leads to our molecules being represented as an N X N sparse matrix.
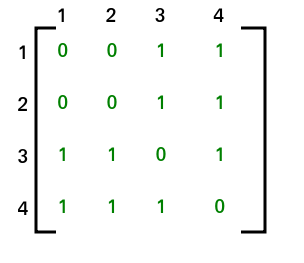
Self-looping was added to all adjacency matrices. Self-looping messages have shown to be useful in deep learning for graph neural networks; this ensures that the node properties affect its own node when training a model. An example of what self-looping messages look like can be seen in the following figure, where n1 and n3 have self-looping.
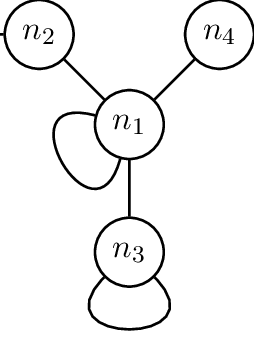
Since we’re interested in more than just the connectivity of our molecules, we must incorporate the chemical information into these feature decompositions for meaningful features, which leads us to the next section
Vocabulary Building
Now that we’ve covered the math decomposition section, we’ll dig into how the vocabulary was decomposed. We mentioned in earlier that the following properties were being tracked for each molecule during the decomposition.
Chemical Properties
- Aromaticity/Rings
- Bond Type (Single, Double, Triple)
- Chirality
- Tertiary/Quaternary Atoms
- Bond Isomerism (Cis/Trans/None)
- Valence
Using prior knowledge, I decided this would be a sufficient list for uniquely identifying molecules and substructures. As a result, a unique dictionary structure was created for each SMILES object using these properties. Each dictionary holds crucial information such as atom-to-index key-value pairs, bond connectivity, and numerical indices assigned by RDKit to indicate all of the other properties stated above.
All the indices for these properties were then converted into strings that summarized the atom, bond type, chirality, atom degree, bond isomerism, and valence. An example of some extracted dictionary entries can be seen below.
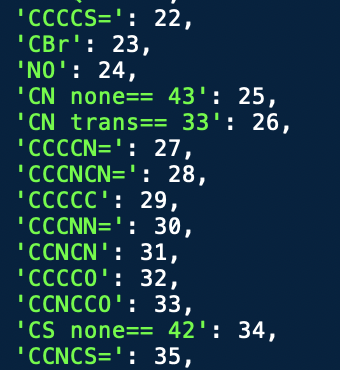
The original dataset used to create the first embeddings in this dictionary was the Zinc250K dataset from Kaggle. From the 250,000 entries, 369 unique atomic structures were identified. Since I knew these embeddings wouldn’t cover the full chemical space, I exported this vocabulary as a JSON file. This would serve as the starting point for a tokenizing algorithm.
After copying the original embeddings into a separate file, functionality was added to dynamically update the token dictionary. To ensure that the algorithm was correctly identifying structures, I made a couple of functions that would draw the RDKit structures based on the vocabulary token given in XX. The following is an example of the token given and the structure returned; in this case, I requested a carbon-oxygen double bonded with “none” isomerism and a valence of 4 and 2 for the carbon and oxygen respectively.
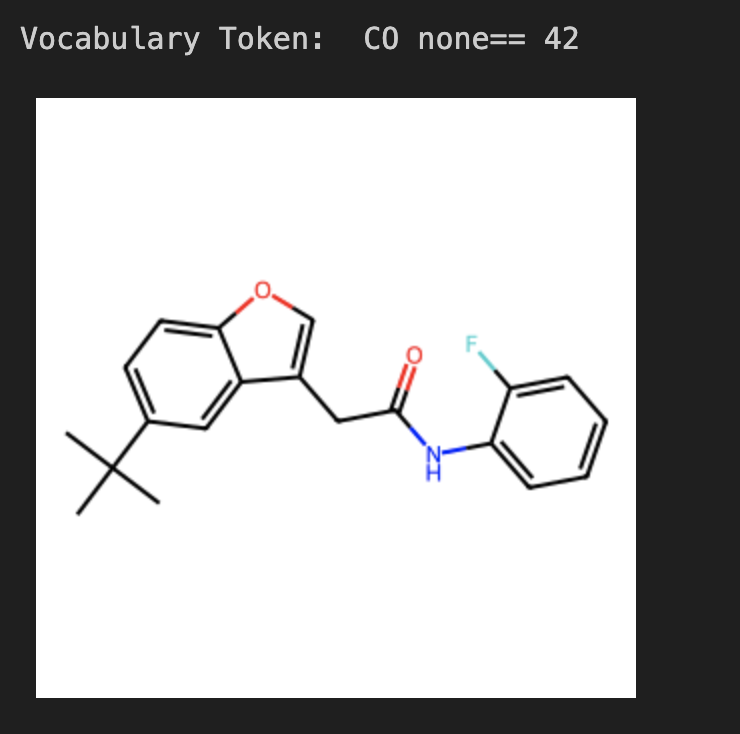
Another verification of this process was the visualization of the junction tree that was generated, more specifically the maximum spanning tree over the junction tree structure that was generated. A maximum spanning tree does not have loops, so that’s the first visual verification of the structure. The next verification is assuring that all nodes are connected, and no errors occurred in creating these structures. To do this, we assure that adjacent nodes have at least one shared atom, and adjacent ring structures should have two shared atoms.
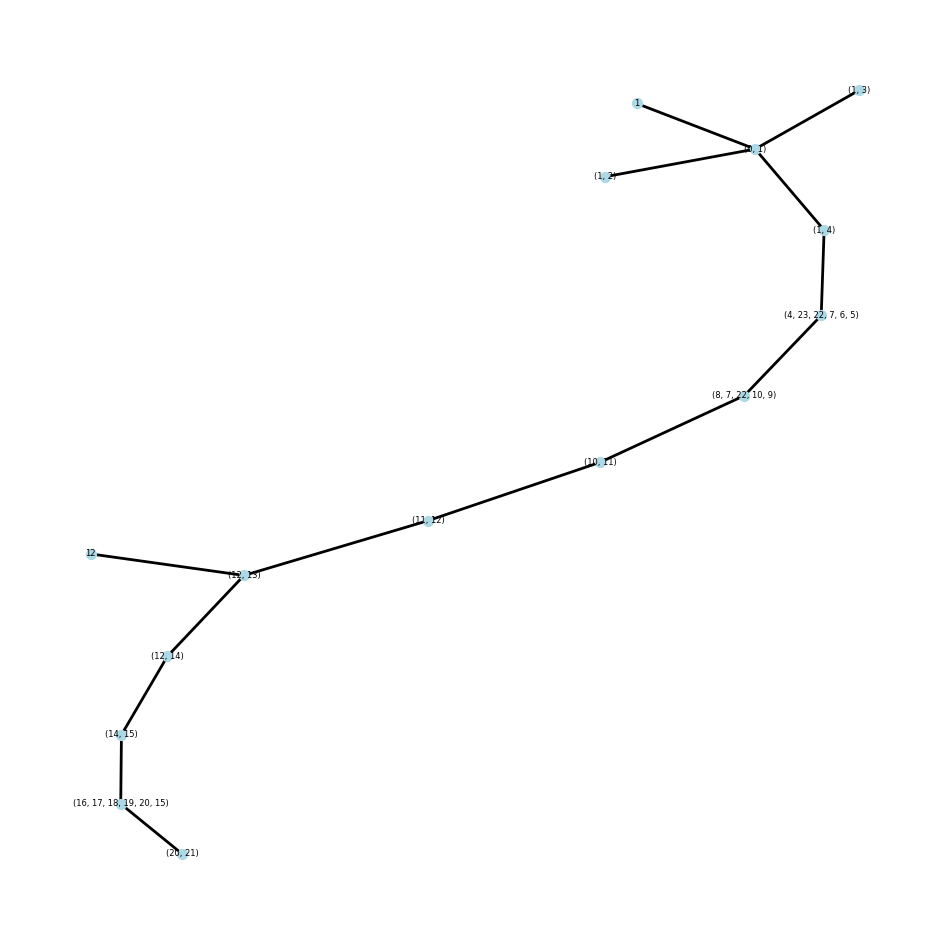
So after these two visual verifications on multiple structures, confidently I can say the tree decomposition algorithm has worked.
Disclaimer
DeepChem offers a variety of beddings that are compatible with graph nueral networks. I learned about this following the creation of this algorithm. I still think it was invaluable to go through this process myself to understand what’s going on under the hood.